COPD is defined
by the Global Initiative for Chronic Obstructive Lung Disease (GOLD) as “a
common preventable and treatable disease, is characterized by persistent
airflow limitation that is usually progressive and associated with an enhanced
chronic inflammatory response in the airways and the lung to noxious particles
and gases. Exacerbations and comorbidities contribute to the overall severity
in individual patients” (GOLD 2011).
This progressive worsening of lung function is caused by narrowing
of the small airways due to increased mucus production, the proliferation of the
smooth muscle cells and fibrosis. This is also accompanied by the destruction
of the alveoli structure (emphysema) and chronic bronchitis (Fig. 1)(Barnes 2007).
Weight loss, nutritional abnormalities and skeletal muscle dysfunction are also
recognised symptoms of COPD (Vogelmeier and Bals 2007). Physical manifestations
of the disease include breathlessness (dyspnea), chronic cough with/without
sputum, wheezing and intolerance to exercise (Qaseem, Wilt et al. 2011).
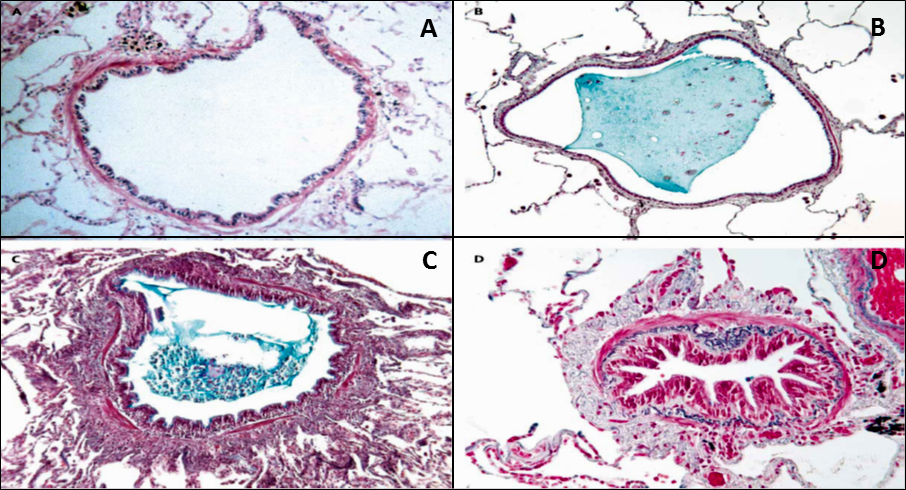
Figure 1.: Histopathology of COPD small airways (A) Normal airway with an open lumen (B) Small airway lumen filled with mucus (C)Acute inflammation and mucus surrounded by thick airway wall (D) Chronic inflammation: fibrosis and hyperplasia of smooth muscle cells accompanied by fibrosis resulting in the obstruction of the airway. Adapted from Hogg (2004).
Epidemiology and prevalence
COPD is ranked as the fifth most common cause of death
globally and predicted to be the fourth leading killer by 2030. It is just
below ischemic heart disease, cerebrovascular disease, and HIV/AIDS (Mathers
and Loncar 2006). It is believed that the lifetime risk of developing COPD is
28% by age 80 (Gershon, Warner et al. 2011). The World Health Organisation
(WHO) projected that it could become the 3rd biggest cause of
mortality by 2020 worldwide (Fig. 2) (Buist, McBurnie et al. 2007). The
prevalence of COPD among the world population counts for about 10%. According
to WHO, there are more than 64 million people affected by COPD (Mathers and Fat
2008). However, this is underestimated and the actual number is higher than 600
million (Sin and Vestbo 2009). Most COPD
patients are either misdiagnosed or unrecognised even long after being severely
disabled (Barnes 2007; WHO 2008). The rate of COPD has increased in the
affluent countries in comparison with the developing countries mainly due to the ageing population and better management of other major illnesses such as cancer
and cardiovascular diseases (Nowak, Berger et al. 2005). One-quarter of the general
adult population age 40 or above have mild airflow obstruction (Buist, McBurnie
et al. 2007; Mannino and Buist 2007; Menezes, Perez-Padilla et al. 2008). For
example, there are estimated 12.2
million (14.3%) people aged ≥ 40 suffers from mild airflow limitation in the
five major cities of Latin American countries (Brazil, Mexico, Uruguay, Chile
and Mexico) (Menezes, Perez-Padilla et al. 2008). Epidemiological studies have
shown that the prevalence of COPD in the USA has doubled between 1979 and 2002 (Decramer,
Janssens et al. 2012). It affects 5% of the adult population and costs the economy
$49.9 billion per annum (Qaseem, Wilt et al. 2011). In Canada, a 2% increase in the numbers of
females with COPD was observed between 1996 and 2007 and 9.5% of the Canadian population
suffer from COPD (Gershon, Wang et al. 2010).
There are still many parts of the world, for example, Africa and Asia,
from which detailed epidemiological data are yet to be reported (Soriano and
Rodriguez-Roisin 2011).
According to the Department of Health (DoH), 25,000 people die
from COPD a year in England and Wales. COPD accounted for 4.8% of all deaths in
England between 2007 and 2009. The DoH reveals that around 835,000 people
suffer from COPD in the UK and an estimated 2.2 million (13% of the population of
England) aged 35 and above remain undiagnosed (Department of Health 2011). COPD
also places an enormous financial burden on the health system and economy as a
direct result of hospitalisation and time lost from work (Barnes 2007; Gruffydd-Jones
2008). It has been reported that GP consultation and hospitalisation has
increased for COPD compared with cardiovascular diseases, resulting in a
greater than 3-fold increase in healthcare costs (Barnes 2007). An estimated
cost for COPD is over £800 million annually in addition to £2.7 billion in lost
working days in the UK alone (Department of Health 2010). It is clear that COPD
poses a serious problem socially and economically that needs to be addressed.

Figure 1.2: World map of estimate
mortality rate caused by COPD in
male patients per 100,000 (WHO updated 2004) (Mathers and Fat 2008; Bhome 2012).
Aetiology
Tobacco smoking is the primary risk factor linked to COPD in
prosperous Westernised countries and environmental pollution, especially indoor
biomass smoke, is associated in the developing world (Decramer, Janssens et al.
2012). Cigarette smoke (CS) is a complex mixture of noxious particles,
chemicals and reactive oxidant species (ROS) (Rahman and Adcock 2006; Min,
Bodas et al. 2011). Physical barriers and the immune system protect the human lung
from harmful environmental agents such as pollution, bacteria, viruses and
fungi (Nikota and Stampfli 2012). However, continuous cigarette smoking affects
these barriers by increasing epithelial permeability and impairing mucociliary
clearance (van der Vaart, Postma et al. 2004; Forteza, Casalino-Matsuda et al.
2012). In addition, both COPD and
smokers are susceptible to respiratory infection, which is the main cause of
exacerbations in COPD patients especially during later stages of the disease
(GOLD III-IV) (Arcavi and Benowitz 2004; Churg, Cosio et al. 2008).
Not all smokers develop COPD and there have been reports of
COPD in non-smokers. A number of risk
factors have been identified, other than cigarette smoking, such as age,
occupational exposure to organic dust, asthma, respiratory infection, biomass
fuel combustion and poor socioeconomic status (Fig. 3) (Salvi and Barnes 2009;
Clancy and Nobes 2012; Forteza, Casalino-Matsuda et al. 2012). In addition,
susceptibility to COPD is also influenced by genetic risk factors. It is long
known that deficiency in alpha1-antitrypsin due to a mutation in the gene
serpin peptidase inhibitor, clade A, member 1 (SERPINA1) leads to COPD and accounts for 1-2% of COPD cases (Kukkonen,
Tiili et al. 2011; Clancy and Nobes 2012).
Recent studies of genomic-wide association (GWA), that
examine the association between common genetic variation and associated phenotype
between groups of individuals, have
identified a number of candidate genes that are associated with COPD (Berndt,
Leme et al. 2012; Bosse 2012; Foreman, Campos et al. 2012). Among these, COPD
patients show a correlation between lung function and single nucleotide
polymorphisms (SNPs) in three loci (GSTCD,
TNS1 and HTR4) (Soler Artigas, Wain et al. 2011). GSTCD gene encodes for glutathione s-transferase C-terminal
domain-containing protein. GSTCD protein catalyses glutathione (GSH) binding to
oxidative stress products and regulates lipid mediators (prostaglandins and
leukotrienes); thereby preventing cellular damage (Hayes, Flanagan et al. 2005).
GSH is an antioxidant that donates an electron (H++e-) to
unstable molecules such as ROS to become oxidised glutathione disulphide (GSSG)
(Flohe 2012). TNS1 gene encodes for tensin-1 adhesion protein that shows binding
preference to actin with SH2 (Src homology 2) domain. Tensin-1 is associated
with cell signalling and migration (Hall, Balsbaugh et al. 2010). HTR4 gene encodes for 5-hydroxytryptamine
4 (5-HT) receptor which is a member of G-protein-coupled receptors (GPCRs) and
expressed in neurones and epithelial type cells (Ghavami, Stark et al. 1999; Wilk,
Shrine et al. 2012). GPCRs are generally expressed in phagocytes for chemokines
and chemoattractants (Sun and Ye 2012). Similarly, BICD1 (Bicaudal D homology 1) is another gene that has been
studied in telomere shortening and cellular senescence (Savale, Chaouat et al.
2009). Leukocytes from COPD patients have reduced telomere lengths compared
with healthy smokers and non-smokers (Savale, Chaouat et al. 2009). This
premature reduction is associated with oxidative stress and inflammatory damage
(Kawanishi and Oikawa 2004). SNP analysis of BICD1 reveals genetic
variation between emphysematous patients and healthy control (Kong, Cho et al.
2011). These loci may provide some
explanation to the underlying cause of COPD but it is ultimately the interaction
between nature (environmental risk factors) and nurture (genotype) that leads
to COPD (phenotype) (Ober, Butte et al. 2010).
Figure 3: Risk factors associated with COPD. Tobacco smoking is the primary risk factor for COPD in about 90% of cases. However, the susceptibility of an individual to COPD also depends on other environmental and genetic risk factors (Salvi and Barnes 2009; Berndt, Leme et al. 2012). Depiction obtained and modified from the Global Initiative for Chronic Obstructive Lung Disease (2011).

Pathophysiology of COPD
COPD is a generic term to describe a number of diseased
conditions including emphysema, chronic bronchitis and small airways disease.
Each condition has its unique clinical features but they all contribute to the
continued restriction of airflow throughout respiratory airways (Barnes 2007; Vestbo,
Hurd et al. 2012). The normal function of the lung is to provide a continuous
supply of oxygen and remove carbon dioxide. It is achieved through simple
diffusion which occurs between alveoli and blood. This exchange mechanism is
vital for survival (Clancy and Nobes 2012). Lung function decreases with age
but it is accelerated in COPD patients due to cigarette smoke (CS) (Fig. 4) (Fletcher
and Peto 1977; Bednarek, Gorecka et al. 2006). Noxious particles in CS cause
irritation of the airways which results in enlargement of the mucus gland, mucus
hypersecretion, ciliary dysfunction and epithelial-cells hyperplasia in the
early stage of the disease. This is accompanied by an increase in goblet cells,
smooth muscle cell mass, and fibrosis (Barnes 2004; Rogers 2005).

Figure 4: The effect of
cigarette smoking on lung function.
Lung function decreases with senescence, however, this is rapidly decreased in COPD patients. Adapted from Fletcher and Peto (1977) and Bednarek, Gorecka
et al. (2006).
CS and other irritants trigger an inflammatory reaction in the
small airways, lung parenchyma and elsewhere in the body (systemic) (Barnes
2008; Voelkel, Gomez-Arroyo et al. 2011). Inflammation occurs not only in COPD
patients but also in healthy smokers (Willemse, Postma et al. 2004). However,
the degree of intensity varies depending on individual susceptibility, genetics
and the magnitude of CS exposure (Dewar and Curry 2006). Persistent
inflammation and ongoing repair of the airways leads to remodelling involving a decrease in airway lumen and an increase in the surrounding tissues (Fig. 5).
This remodelling is accompanied by excess mucus in the airways, causing airflow
obstruction (Decramer, Janssens et al. 2012).
In addition, chronic inflammation and increase proteinases such as
elastase and matrix metalloproteinases (MMPs) released from macrophages and
neutrophils cause the disappearance of surrounding alveoli, leaving behind
abnormal enlarged airspace-termed emphysema. This has an impact on the O2
and CO2 exchange mechanism (Min, Bodas et al. 2011; Voelkel,
Gomez-Arroyo et al. 2011).

Figure 5: Comparison between
healthy and COPD airways. (A) Normal airway: Large lumen and
thin structural walls (B) COPD airway: Lumen is narrowed and surrounded
by inflamed tissues (Decramer, Janssens et al. 2012).
COPD and asthma share some similar clinical features but
there are major differences. Both are inflammatory disorders of the respiratory tract
and cause narrowing of the airways. However, asthma often starts in childhood
and inflammation is localised to larger airways, whereas, COPD develops in the age
above 40 and is associated with inflammation of small airways (Barnes 2008; Barnes
2008). The inflammatory response in asthma is mainly triggered by allergen and
mediated by dendritic cells, eosinophils, activated mast cells, CD+4
T cells (Th2 cells). In contrast, COPD is associated with CS and recruit
macrophages and neutrophils and CD+ 8 T cells at the site of
inflammation (Fig. 6) (Barnes 2008; Zanini, Chetta et al. 2010). Steroids are the most effective
anti-inflammatory drugs and are frequently used in asthma. However, they are
ineffective in most patients with COPD (Barnes 2006).

Figure 6: The difference
between asthma and COPD. There are some similarities and differences
between asthma and COPD including both histopathological and immunological
changes which are highlighted above.
Adapted and modified from Barnes (2008).
Diagnosis
COPD is diagnosed depending on signs, symptoms and medical
history. However, a spirometry test is frequently used to assess lung function.
Spirometry test shows an irreversible decrease in forced expiratory volume in 1
second (FEV1) and the ratio of FEV1 to forced vital
capacity (FEV1/FVC) (Mannino and Buist 2007; 2011). The severity of
the disease is classed into 4 categories depending on FEV1/FVC ratio
(Table 1.). X-ray and quantitative computed tomography (CT) are also utilised
to evaluate lung function and structure (Mets, de Jong et al. 2012). The chest CT scans assess lung density, which
is proportional to lung airspace enlargement otherwise known as emphysema (Berndt,
Leme et al. 2012).
|
Classification of COPD severity
|
Classification based on post-bronchodilator lung function
|
|
GOLD 1 (mild)
|
FEV1/FVC
<0·70 and FEV1 ≥80% predicted
|
|
GOLD 2 (moderate)
|
FEV1/FVC
<0·70 and 80% >FEV1 ≥ 50% predicted
|
|
GOLD 3 (severe)
|
FEV1/FVC
<0·70 and 50% >FEV1 ≥ 30% predicted
|
|
GOLD 4 (very severe)
|
FEV1/FVC
<0·70 and FEV1 <30% predicted or FEV1 <50%
predicted plus chronic
respiratory failure
|
The multifaceted nature of COPD has proved challenging to
develop a treatment that can target all the components of the disease (Barnes
2010). Currently, combinations of pharmacological and non-pharmacological
strategies are used in the management of COPD. Pharmacological options include
bronchodilators, inhaled corticosteroids (ICS), combination therapy and
long-term oxygen therapy (LTOT). Non-pharmacological interventions are smoking
cessation, pulmonary rehabilitation, mechanical ventilation and
lung-volume-reduction surgery (LVRS) (Hanania, Ambrosino et al. 2005). Among these treatments, bronchodilators and
ICS are frequently prescribed (Barnes and Stockley 2005).
Bronchodilators are used for symptomatic relief, which
consists of short-acting 2-agonists (Salbutamol) and long-acting
(Salmeterol and Formoterol) or anti-muscarinic drugs like Tiotropium. They
improve FEV1 in some patients, but, the effect on lung function,
reduction in inflammation and exacerbation remain poor (Hanania, Ambrosino et
al. 2005; Barnes 2010; Bhome 2012). ICS are
also used either alone or in combination with bronchodilators in the management
of inflammation in COPD patients. However,
they provide little or no benefits to COPD patients (Hakim, Adcock et al. 2012).
It has minimal significant effects on
some of the key inflammatory mediators such as CXCL8/IL-8, tumour necrosis
factor-alpha (TNF-a) and MMPs (Barnes 2007). ICS and antibiotics are
recommended by GOLD for symptomatic treatments in patients with exacerbation
and whose FEV1<50% of predicted (GOLD 2011; Mackay and Hurst 2012). Glucocorticoids are the most effective and
widely used therapy in many inflammatory and immune diseases with exception
of COPD (Barnes and Adcock 2009). This lack of response is partially explained
by cigarette smoke and oxidative stress, reducing HDAC2 activity and expression
as well as impairing GR translocation (Barnes and Adcock 2003; Barnes 2009).
However, it may also be due to other epigenetic changes, including alteration
in DNA and histones status, under oxidative stress (Adcock, Tsaprouni et al.
2007; Adcock, Chou et al. 2009).
Disclaimer: The
information in this blog is for general purposes only. A patient must seek clinical advice and treatment from a qualified physician.
The information may not be up to date and should not be used as accurate
and reliable. Any reliance you place on such information is therefore strictly
at your own risk.
Selected references for further reading
Barnes, P. J. (2007). "Chronic obstructive pulmonary
disease: a growing but neglected global epidemic." PLoS Med 4(5): e112.
Barnes, P. J. (2010). "New therapies for chronic
obstructive pulmonary disease." Med Princ Pract 19(5): 330-338.
Barnes, P. J. (2012). "Development of New Drugs for
COPD." Curr Med Chem. Sep 3. [Epub ahead of print] PMID: 22963554
Hogg, J. C. (2004). "Pathophysiology of airflow
limitation in chronic obstructive pulmonary disease." Lancet 364(9435): 709-721.
Hogg, J. C. (2006). "Why does airway inflammation
persist after the smoking stops?" Thorax 61(2): 96-97
Department of Health
(2011). "An outcomes strategy for people with chronic obstructive
pulmonary disease (COPD) and asthma in England." from http://www.dh.gov.uk/en/Publicationsandstatistics/Publications/PublicationsPolicyAndGuidance/DH_127974.